What are AAVs? and what makes them good candidates for gene therapy?
By: Christopher Reardon, Stephen Malina, and Eryney Marrogi
Introduction
In the first part of our three part series introducing AAV as a gene therapy vector, we talked about basic AAV vector biology. In this post, we’re going to take a step back to answer the question of “Why AAV?” and look at some opportunities in the AAV engineering space.
Why AAV?
Viral vectors are one of the three main classes of gene therapy delivery vehicles. The other two are lipid nanoparticles (LNPs), now famous for their roles in mRNA vaccines, and plasmid electroporation. Relative to these other options, viral vectors have the advantage of higher delivery efficiency, better targeting, and a long clinical history starting with their use in vaccines and more recently, in gene therapy.
Three main viral vectors exist within the viral vector gene therapy landscape [1]: lentiviruses, adenoviruses, and AAVs. With respect to their use in gene therapy, we can assess viral vectors in terms of their safety, production robustness and scalability, ability to target specific tissues (tropism), and packaging capacity. Each makes various trade-offs with respect to these characteristics.
Lentiviruses are single-stranded RNA viruses originally derived from natural retroviruses. When used for gene therapy, lentiviruses have the advantage of a large package capacity (~9 kilobases) but present challenges related to genome integration [2]. Adenoviruses are double-stranded DNA viruses best known for causing the common cold. The newest generation of adenoviruses can package large transgenes of length up to ~36kb and combine high transduction efficiency with scalable viral production. However, they tend to trigger strong immune responses in humans, which limits their applicability for gene therapy applications, particularly in immunocompromised patients [1].
Compared to adenovirus and lentivirus, AAV offers a low immune profile, relative safety, long-lasting expression in non-dividing cells, and a long scientific and clinical history. Two AAV-based gene therapies — Luxturna® and Zolgensma® — have already been approved by the FDA, and many more [4] are in various stages of clinical trials. However, AAV can only package ~4.7 kilobases of linear single-stranded DNA, meaning packaging full genes encoding large proteins (such as dystrophin) currently requires gene size optimization and/or spreading a gene across multiple vectors.
Opportunities in AAV engineering
While it’s impressive how well natural variants of AAV work for gene therapy, they lack certain features that would make them even more useful. As mentioned above, they can only package ~4.7kb, so fitting transgenes larger than 4.7kb into a vector requires clever, but error-prone, strategies. In addition, although different natural variants do weakly target certain tissue types, this targeting has not been optimized for precision. Finally, although the AAV capsid does not trigger a strong immune response, natural immunity to it can develop, which makes repeated dosing difficult by increasing the risk of potential side effects and reducing dosing efficiency.
To overcome these challenges, researchers have focused on improving the AAV capsid protein and designing promoters that modulate expression of the delivered gene. The first of these, capsid design, involves modifying the capsid gene to change its function. Existing work in this category has focused on:
- Increasing packaging capacity beyond ~4.7kb,
- Targeting (and de-targeting) specific tissues such as the brain [3] or liver, and
- Improving the ability of the capsid to evade the immune system, even after repeated administration.
However, any approach to solving these problems must grapple with narrowing the space of possible capsid variants. Our goal in design is to find a set of promising variants that’s small enough to test in vitro/vivo (scale) while maximizing the fraction of capsids in this set which are viable (efficiency). However, achieving this poses a substantial challenge given that even a single VP1 monomer has 20^735 possible variants. Traditional approaches typically optimize for one of scale vs. efficiency.
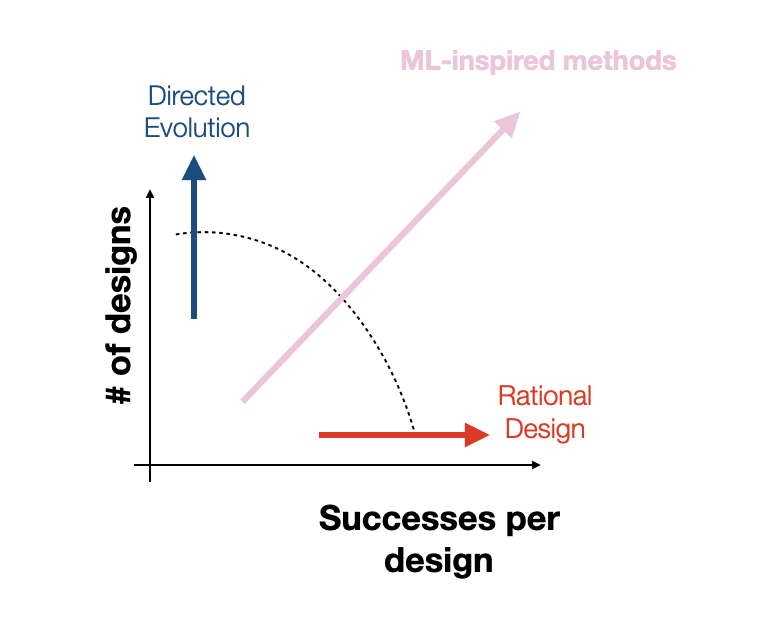
For instance, directed evolution and unbiased random mutagenesis approaches test as many as 10^10 variants, which, while large, amounts to a drop in the bucket of an incredibly vast space that is many orders of magnitude larger. Additionally, in the case of the “massive” random mutagenesis libraries, the majority of tested variants end up nonviable, amounting to an extremely inefficient search. On the other hand, rational design approaches make targeted mutations to small sub-regions of the capsid based on hard-won biological knowledge, increasing efficiency at the cost of reducing scale. Rational design approaches excel at discovering small local perturbations to the capsid that increase fitness but lack the scale and throughput to identify mutations that take advantage of unexpected, potentially nonlinear effects of mutations to disjointed capsid sub-regions. At Dyno, we hope to find the right balance of efficiency and scale by combining machine-guided design with high-throughput screening. Although our high-throughput screens typically test between 10^4 and 10^5 variants per library, as we’ve shown in previous work, our machine learning models increase the efficiency of these libraries by vastly increasing the proportion of viable variants in the library [5, 6]. By directly modeling the relationship between sequence and function, we can further guide our search towards capsids that not only produce but also have optimized desired properties.

Conclusion
Taking a step back, it’s important to put AAV engineering in context with respect to the larger project of improving our ability to control cellular outcomes and behavior. Allowing ourselves to speculate a little, we can think of AAV as an impressive but imperfect starting point for a future generic in vivo gene delivery system. Looked at through this lens, any deficiency of AAV that prevents it from safely delivering genes (repeatedly) in all patients with arbitrary tissue and cell type specificity in a low dosing regime presents an opportunity for AAV engineering.
As we said in our prior post, this wouldn’t be a company blog post if we didn’t mention that we’re currently hiring for a range of technical and non-technical roles. If you’re excited about working with us, please apply and for the authors’ sake, mention that our blog post played a role in getting you excited about Dyno!
Special thanks to: Adam Poulin-Kerstein, Alex Brown, Cherry Gao, Eric Kelsic, Heikki Turunen, Jeff Gerold, and Sam Sinai for helpful comments.
[1]: Bulcha, J.T., Wang, Y., Ma, H. et al. Viral vector platforms within the gene therapy landscape. Sig Transduct Target Ther 6, 53 (2021). https://doi.org/10.1038/s41392-021-00487-6
[2]: Milone, M.C., O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 32, 1529–1541 (2018). https://doi.org/10.1038/s41375-018-0106-0
[3]: Ravindra Kumar, S., Miles, T.F., Chen, X. et al. Multiplexed Cre-dependent selection yields systemic AAVs for targeting distinct brain cell types. Nat Methods 17, 541–550 (2020). https://doi.org/10.1038/s41592-020-0799-7
[4]: Kuzmin, Dmitry A., et al. “The clinical landscape for AAV gene therapies.” Nature reviews. Drug Discovery (2021).
[5]: Sinai, Sam, et al. “Generative AAV capsid diversification by latent interpolation.” bioRxiv (2021).
[6]: Bryant, D.H., Bashir, A., Sinai, S. et al. Deep diversification of an AAV capsid protein by machine learning. Nat Biotechnol 39, 691–696 (2021). https://doi.org/10.1038/s41587-020-00793-4